

Ein progressiver Nierenfunktionsverlust ohne auslösendes Agens, ein blandes Urinsediment, weder Proteinurie noch Albuminurie. Jedoch als weitere Befunde interstitielle Fibrose, tubuläre Atrophie, negative Immunfluoreszenz für Komplementfaktoren und Immunglobuline. Dazu eine Häufung von "Gichtnieren" in der Familienanamnese – selten, aber typisch für eine ADTKD.
Die autosomal-dominante tubulointerstitielle Nierenerkrankung (ADTKD) zeichnet sich durch Mutationen in verschiedenen Genen aus. Es wird vermutet, dass die ADTKD eine der häufigsten monogenetischen Formen einer Nierenerkrankung darstellt. Ca. 5 % aller monogenetischen Ursachen einer chronisch funktionellen Nierenerkrankung sind auf eine ADTKD zurückzuführen (1).
Die häufigsten (ca. 50 %) aller ADTKD werden durch Mutationen im UMOD-Gen, das für Uromodulin codiert, oder am Gen MUC1, das für Mucin-1 codiert, verursacht (1).
Des Weiteren kommen unter anderem Mutationen im Gen für den Transkriptionsfaktor 2, auch als hepatocyte nuclear factor 1-beta (Hepatozyten-nukleärer Faktor 1-beta, HNF1B) bezeichnet, und für Renin (REN) vor.
Die Erkrankungen wurden vor ihrem Zusammenfassen unter dem Oberbegriff der ADTKD unter verschiedensten Bezeichnungen als einzelne Krankheiten geführt, unter anderem als medullär-zystische Nierenerkrankung Typ II, familiäre juvenile hyperurikämische Nephropathie oder Uromodulin-assoziierte Nierenerkrankung. Alle diese Bezeichnungen waren jedoch eher irreführend und die KDIGO fasste diese Erkrankungen daher unter dem Terminus "autosomal-dominante tubulointerstitielle Nierenerkrankung" zusammen mit einer genbasierten Subklassifikation.
All diesen Erkrankung ist gemein, dass sie eine progressive tubulointerstitielle Fibrose aufweisen bis zur terminalen Nierenerkrankung.
Bemerkenswert ist hierbei, dass alle betroffenen Gene in tubulären Zellen im intermediären oder distalen Nephron exprimieren. Uromodulin (UMOD) kommt im dicken aufsteigenden Teil der Henleschen Schleife vor. Die klinische Manifestation von Mutationen im UMOD-, Renin- und Mucin-1-Gen ist dabei ausschließlich auf die Niere beschränkt. Im Gegensatz hierzu weist Hepatozyten-nukleärer Faktor 1-beta (HNF1B) auch extrarenale Manifestationen auf. Eine Minderheit dieser Patienten weist jedoch auch eine progressive interstitielle Fibrose der Niere auf.
Im Gegensatz zu der ADPKD spielt eine Zystenbildung bei der ADTKD keine pathologische Rolle.
Typische klinische Charakteristika einer ADTKD
Typische Charakteristika für die ADTKD sind eine positive Familienanamnese mit der Beteiligung von mehreren Familienmitgliedern in verschiedenen Generationen. Hierbei werden oft nicht alle betroffenen Familienmitglieder diagnostiziert, da sie bereits zuvor versterben. Die Penetranz der verschiedenen ADTKD-Typen beträgt bis zu 100 % der Patienten, wenn sie das Alter erreichen, in dem sich ihre Erkrankung manifestiert.
Aber es finden sich auch de-novo-Mutationen, vor allem wenn HNF1B betroffen ist.
ADTKD-Patienten und -Patientinnen ist gemein, dass sie ein blandes Urinsediment aufweisen. In Einzelfällen kann eine Mikrohämaturie vorkommen, Proteinurie oder Albuminurie sind jedoch meist nicht vorhanden. Initial normwertige Nierengrößen können sich im Verlauf der Erkrankung zu Schrumpfnieren entwickeln. Zystennieren können vorkommen, jedoch spielen sie pathognomonisch keine Rolle. Eine arterielle Hypertonie gehört im Frühstadium nicht zur ADTKD, beim weiteren Fortschreiten der Erkrankung kann eine moderat erhöhte arterielle Hypertonie jedoch vorkommen.
- Interstitielle Fibrose
- Tubuläre Atrophie
- Verdickung und Lamellierung der tubulären Basalmembran
- Mögliche tubuläre Dilatation (Mikrozysten)
- Negative Immunfluoreszenz für Komplementfaktoren und Immunglobuline
ADTKD-UMOD-Mutation
Die erste berichtete heterozygote UMOD-Mutation wurde 2002 in 4Familien diagnostiziert. Hier konnte ein autosomal-dominanter Erbgang nachgewiesen werden. Diese Patienten wiesen in der Adoleszenz eine Hyperurikämie und Gicht auf. Aktuell sind etwa 135Mutation diagnostiziert worden. Die Penetranz einer UMOD-Mutation liegt bei nahezu 100 %. Es gibt nur wenige heterozygote Formen, bei denen die Patienten keine klinische Manifestation aufweisen (1).
- Patienten mit einer UMOD-Mutation weisen charakteristischer Weise eine verminderte Urat-Exkretion auf.
- Die Hyperurikämie beginnt meistens in der Kindheit als erstes Symptom vor einer renalen Erkrankung.
- Patienten mit UMOD-Mutation weisen in ihrer Krankengeschichte häufig Gichtanfälle auf.
- Die Gicht kommt oft bei vielen Mitgliedern einer Familie vor, sie muss aber nicht zwingend bei allen vorhanden sein. Sie kann ein Schlüssel zur Diagnosestellung sein.
- Die Gicht tritt oft bereits im jugendlichen Alter und vor allem bei männlichen Familienmitgliedern auf.
- Ungefähr 60 % der Männer und 45 % der Frauen mit einer UMOD-Mutation entwickeln eine Gicht. Das mittlere Alter beträgt dabei bei Männern 29 Jahre und bei Frauen 30 Jahre. In etwa 25 % tritt die Gicht bereits vor dem 25. Lebensjahr auf.
- Das klinische Manifestationsalter einer terminalen Nierenerkrankung bei UMOD-Mutation liegt zwischen dem 25. und dem 70. Lebensjahr, gelegentlich aber auch im noch höheren Lebensalter.
- Etwa 1 % aller Dialysepatienten weisen eine UMOD-Mutation auf.
ADTKD-MUC1-Mutation
Patientinnen und Patienten mit einer MUC1-Mutation weisen ebenso wie diejenigen mit einer UMOD-Mutation eine variable Progression der CKD auf. Sie erreichen mit durchschnittlich dem 45. Lebensjahr schließlich eine terminale dialysepflichtige Nierenerkrankung, wobei die CKD selten vor dem 20. Lebensjahr beginnt. Die CKD ist bei der MUC1-Mutation die einzige primäre klinische Manifestation der Erkrankung (2).
HNF1B-Mutation
Patientinnen und Patienten mit Mutation von HNF1B weisen im Gegensatz zu MUC1-Patienten häufig eine erste renale Manifestation bereits vorgeburtlich oder in der Kindheit auf. Diese Patienten erreichen im Durchschnitt die terminale Nierenerkrankung im Alter von 7 bis 48 Jahren.
Dialysepflichtigkeit bei UMOD-, MUC1- und HNF1B-Mutationen
Das Alter, in dem Patientinnen und Patienten mit einer dieser Mutationen eine Dialysetherapie benötigen, liegt zwischen dem 20. und dem 80. Lebensjahr, meistens jedoch in dem Zeitraum zwischen dem 49. und 54. Lebensjahr. Die Progressionsrate ist variabel.
REN-Mutation
Eine Besonderheit weisen Patienten mit ADTKD-REN-Mutation auf. Sie entwickeln meist in der Kindheit eine Anämie. Diese scheint unabhängig von der glomerulären Filtrationsrate und somit der Nierenfunktion zu sein. Die genauen Ursachen der kindlichen Anämie sind bis heute ungeklärt. Der durchschnittliche Hb-Wert liegt zwischen 8 und 11 g/dl. In der Pubertät verschwindet die Anämie meistens. Im weiteren Verlauf der Erkrankung mit zunehmendem Nierenfunktionsverlust kann sie jedoch erneut auftreten.
Patienten mit einer REN-Mutation weisen zusätzlich in der Kindheit oft einen Hypoaldosteronismus mit begleitend sinkender GFR auf. Klinisch manifestiert sich dieser in einem niedrigen Blutdruck, Übelkeit und einer Gedeihstörung. Zudem zeigt sich laborchemisch eine Hyperkaliämie. Alle Patienten weisen einen niedrigen Plasmareninspiegel auf. Die verminderte GFR könnte in der verminderten Reninproduktion begründet sein. Diese Patienten neigen zu einem akuten Nierenversagen im Rahmen von einer Dehydration, vor allem bei viralen Infekten (2).
Wichtige diagnostische Kriterien für eine ADTKD:
- Eine familiäre Disposition mit autosomal-dominantem Erbgang und CKD
- Ein Schlüssel zur Diagnose ist hier wie so oft die Familienanamnese.
- Bei Abwesenheit einer positiven Familienanamnese sind die klinischen Manifestationen und der Befund einer Nierenbiopsie wegweisend.
- Zudem sollte nach einer früh einsetzenden Hyperurikämie oder Gicht gefragt und auf mögliche extrarenale Manifestation geachtet werden (HNF1B).
Diagnostik:
Mittels genetischer Analysemethode NextGenerationSequencing.
Therapie:
- Nach dem ersten Gichtanfall sollte mit Allopurinol oder einer anderen harnsäuresenkenden Therapie behandelt werden.
- Zudem sollte eine strikte purinarme Kost eingenommen werden.
- Eine diuretische Therapie sollte zurückhaltend bei allen Patienten mit einer nachgewiesenen ADTKD angewandt werden, denn sie kann mögliche Gichtanfälle unter Volumenmangel begünstigen.
- Es ist auf eine ausreichende Hydratation bei allen ADTKD-Patienten zu achten.
- Nicht-steroidale Antirheumatika sollten vermieden werden. Vor allem bei Patienten mit ADTKD-REN-Mutation können diese zu einer akuten Verschlechterung der Nierenfunktion beitragen (3).
- Patienten mit REN-Mutation können mit Erythropoetin-stimulierenden Agenzien und Fludrocortison behandelt werden (2).
- Therapie der Wahl ist bei nachgewiesener ADTKD und terminaler Nierenerkrankung eine Nierentransplantation, da hiermit (wie auch bei der ADPKD) die genetische Mutation nicht mehr zum Tragen kommt und somit keine Rezidivgefahr besteht.
Vorsorge:
Vorsorglich sollten alle potenziell betroffenen Familienmitglieder regelmäßig auf andere Risikofaktoren bezüglich einer gravierenden CKD untersucht und behandelt werden. Zudem sollten sie regelmäßig nephrologisch überwacht werden (3, 2).
Patient 1: Vater von Patient 2
- ADTKD bei terminaler Nierenerkrankung, Z.n. Nierentransplantation, primäre familiäre Hyperurikämie
- PTLD im Sinne eines diffusen großzelligen B-Zell-Non-Hodgkin-Lymphoms. Befall des Jejunums und der retroperitonealen Lymphknoten
- Z.n. Ileumperforation mit Peritonitis
- Arterielle Hypertonie
- Hyperlipidämie
- Zwei-Gefäß-KHK: PTCA plus DES im Ramus diagonalis
- Cholezystolithiasis
- Z.n. Neurolyse des Nervus genitofemoralis und Verschluss Hernia inguinalis rechts mit Netzeinlagr
- Z.n. CMV Colitis
- Asthma bronchiale
- Herpes Zoster am Kopf linksseitig
- Bronchitis und Sinusitis mit Leukopenie bei Knochenmarkstoxizität unter Azathioprin in Kombination mit Allopurinol
- Metabolische Azidose a.n.k.
- Pathologische Glucosetoleranzstörung
- Sekundärer renaler Hyperparathyreoidismus
Anamnese:
Die Übernahme des Patienten erfolgte aus der hausärztlichen Praxis. Der Patient ist seit dem 42. Lebensjahr nierentransplantiert und in der Universitätsklinik angebunden. Es besteht eine Immunsuppression mit Azathioprin und Tacrolimus. Aktuell berichtet er über periphere Ödeme, hier ist eine Lymphdrainage geplant. In der kardiologischen Abklärung im Frühjahr2022 zeigten sich keine interventionsbedürftigen pathologischen Befunde, auch nicht im Belastungs-EKG.
Der Patient leidet aktuell unter einer Thrombose des rechten Armes bei Z.n. Port-Anlage auf dieser Seite sowie einer erhöhten Blutbildungsrate. Es wurde dies in diesem Rahmen interpretiert und mit Eliquis behandelt.
Die Erstdiagnose der möglichen "Gichtnierenerkrankung" erfolgte mit dem 12. Lebensjahr nach dem Tod der Mutter. Hier wurde eine erhöhte Harnsäure festgestellt und der Patient entsprechend medikamentös behandelt.
Er berichtet aktuell nicht über urämische Symptome, ebenso keine Angina-Pectoris-Symptomatik.
Z.n. Wunde im Ösophagus unklarer Genese.
Familienanamnese:
Die Mutter verstarb vor 45 Jahren in ihrem 46. Lebensjahr an einer "Gichtniere" im Rahmen eines Coma urämicum mit hochgradig eingeschränkter Nierenfunktion. Dekompensierte Hypertonie und Herzinsuffizienz sowie sekundäre Anämie. Bei der Aufnahme damals war sie somnolent und zeigte ein blassgelbes Hautkolorit. Der Körper war von zahlreichen kleinen aufgekratzten Effloreszenzen an Stamm und Extremitäten bedeckt. An beiden Unterschenkeln bestanden starke Ödeme sowie beidseitige "geschwürige" Veränderungen. In der Umgebung der Ulzerationen fanden sich multiple stecknadelkopfgroße weißliche Knötchen.
Im Labor zeigte sich ein Harnstoff von 300mg/dl und ein Kreatinin von 5,18mg/dl. Der Hb-Wert lag bei 6,0mg/dl.
Unter einer begonnenen Peritonealdialyse zeigte sich initial eine Regredienz der Symptomatik. Sie litt ebenfalls unter einer Arthritis urica sowie einer doppelseitigen pyelonephritischen Schrumpfniere. Im weiteren Verlauf verstarb die Patientin im Rahmen der Urämie.
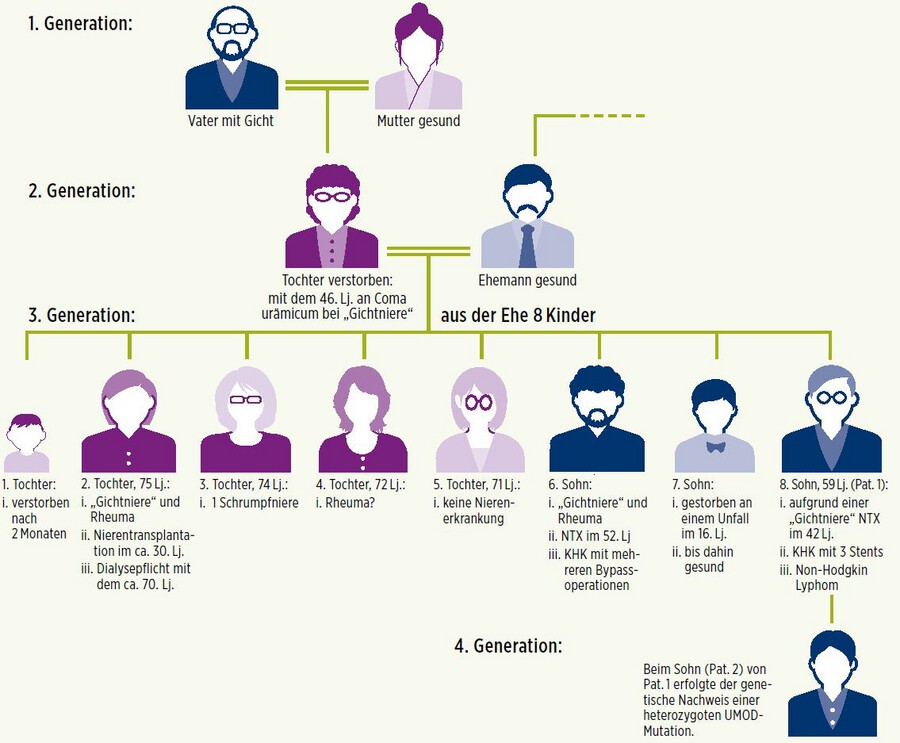
Von den 6 lebenden Geschwistern des Patienten sind 3 nierentransplantiert aufgrund der "Gichtnierenerkrankung" (siehe Stammbaum).
{kind=link}
Der Bruder mit der Nierentransplantation erhielt vor Jahren Bypässe aufgrund einer KHK, der Vater des Patienten verstarb an "Herzasthma" mit dem ca. 71. Lebensjahr.
Der Sohn des Neffen ist kleinwüchsig und zwei Nichten des Patienten haben eine Multiple Sklerose.
Der Patient ist verheiratet und hat einen Sohn (= Pat. 2), bei dem eine ADTKD-UMOD-Mutation genetisch diagnostiziert wurde.
In der persönlichen Anamnese berichtet der Patient über ein Z.n. Medikamentenintoxikation mit Knochenmarksbeteiligung und Blutarmut durch Kombination von Azathioprin und Allopurinol vor zwei Jahren. Hier bekam der Patient entsprechend Blutkonserven.
Er berichtet weder über einen Nikotin- noch einen Alkoholabusus, keine NSAID in letzter Zeit, kein CT mit Kontrastmittel in letzter Zeit. Von Beruf war der Patient Elektrotechniker. Hier kam er nicht mit Giftstoffen in Verbindung, keine HIV- oder Hepatitisansteckung bekannt.
Z.n. rezidivierenden Blutübertragungen, letztmalig vor zwei Jahren. Im Alter von 18 Jahren hatte der Patient eine unklare Leberwerterhöhung mit Ikterus.
In der Systemanamnese berichtet er über eine Gewichtszunahme im Rahmen der erhöhten Cortisontherapie.
Schnarchen und Atemaussetzer sind bekannt bei verlängertem Zäpfchen. Hier ist eine operative Revision geplant, ein Schlafapnoescreening schloss ein Schlafapnoesyndrom aus.
Es bestehen Schmerzen in den Gelenken, vor allem der Arme. Aktuell bestehen periorbitale Ödeme mit Wassereinlagerung im Rahmen der Cortisontherapie, bekanntes Asthma bronchiale unklarer Genese.
Der sporadisch gemessene Blutdruck liegt bei 125/70mmHg, periphere Ödeme wie beschrieben, Refluxösophagitis, Teerstuhl im Rahmen einer Eisensubstitution, normale Trinkmenge.
Neurologische Ausfallerscheinungen mit dem "Einschlafen" der Hände bei bekanntem Karpaltunnelsyndrom bds. sowie HWS-Symptomatik.
Bekannte Tetracyclin-Allergie mit Hauterscheinungen sowie unerwünschte Nebenwirkung von Ibuprofen mit Fieberschüben.
Keine anamnestischen Hinweise auf eine Vaskulitis.
Körperlicher Untersuchungsbefund:
Patient in gutem AZ und adipösem EZ, Größe 166cm, Gewicht 73,0kg, BMI 26,49 kg/m², Blutdruck am linken Oberarm gemessen im Sitzen 155/87mmHg, Puls 74/min.
Bis auf periorbitale Ödeme unauffällig.
Nierenfunktion:
GFR nach CKD-EPI-Kreatinin-Cystatin C: 26,5 ml/min/1,73m2
Beurteilung:
Die Übernahme des Patienten erfolgte bei genetisch nachgewiesener familiärer ADTKD. Der Sohn des Patienten wurde mittels NextGenerationSequencing diagnostiziert.
Die Nierenfunktion zeigt sich mit einer eGFR von 26,5ml/min pro KOF und einem Kreatinin von 2,15mg/dl stabil zu den Vorwerten. Diese liegt seit neun Monaten zwischen einem Kreatinin von 2,27 und 1,99mg/dl sowie einer eGFR von 29,1ml/min pro KOF und einer eGFR von 35,9ml/min/KOF.
Laborchemisch zeigt sich aktuell eine stabile renale Anämie unter Erythropoetintherapie. In Anbetracht des Z.n. Thrombose empfehle ich engmaschige Verlaufskontrollen mit einem maximalen Hb-Wert von 11,5mg/dl, da sonst eine erhöhte Gefahr einer Re-Thrombose besteht unter der Therapie mit Erythropoetin. Aufgrund eines persistenten Eisenmangels trotz oraler Eisensubstitution wurde die Therapie auf orales Eisen-III-Maltol umverordnet. Sollte sich hier keine Verbesserung des Befundes zeigen, wäre eine i.v. Eisentherapie anzuraten. Eine weitere Verlaufskontrolle des Eisenstatus sollte erfolgen. Der Folsäurespiegel zeigte sich therapeutisch unter Substitution.
In der Urindiagnostik zeigte sich eine signifikante Proteinurie bei Nachweis einer moderat erhöhten Albuminurie.
Der weitere Urinstatus war unauffällig.
Auf eine Sonografie des Abdomens sowie die körperliche Untersuchung wurde explizit auf Wunsch des Patienten verzichtet bei vor Kurzem stattgefundenen CT-Abdomen und-Thorax. In diesem zeigten sich unklare Verschattungen, weshalb der Patient zum Pneumologen zur weiteren diagnostischen Abklärung bei bekanntem diffusen großzelligen B-Zell-Non-Hodgkin-Lymphom überwiesen wurde.
In der BGA zeigte sich eine therapeutische metabolische Azidose unter Therapie mit bicaNorm. Diese sollte fortgeführt werden.
Der Knochenstoffwechsel wies einen stabilen sekundären Hyperparathyreoidismusauf. Die Therapie mit Alfacalcidol wurde beendet bei erhöhtem Vitamin-D-Spiegel. Hier sind weitere Verlaufskontrollen des Vitamin-D-Status vonnöten.
Das HbA1c von 6,1 % wies auf eine pathologische Glukosetoleranzstörung unter der Cortisontherapie hin. Hier sind weitere Verlaufskontrollen und gegebenenfalls eine diabetologische Vorstellung indiziert.
Eine Hypertriglyzeridämie ist im Rahmen der pathologischen Glukosetoleranzstörung zu interpretieren und sollte diätetisch behandelt werden.
Die Hypercholesterinämie zeigte sich unter Therapie mit Rosuvastatin nicht therapeutisch. Bei einer GFR unter 30 ml/min/KOFist eine Therapie mit Rosuvastatin kontraindiziert. Hier wurde eine Intensivierung der Therapie mit Atorvastatin 30 mg und ergänzend Ezetimib 10 mg/d begonnen.
Eine Hyperurikämie zeigte sich unter der Therapie mit Benzbromaron therapeutisch.
Aufgrund der hypertensiven Blutdruckwerte wurde die Therapie auf Vocado HCT 40/5/12,5 mg umverordnet. Hier ist eine zeitnahe Kalium- und Kreatininkontrolle geplant.
Insgesamt zeigt sich eine stabile chronisch funktionelle Nierenerkrankung im CKD-Stadium IV bei Z.n. Nierentransplantation im Rahmen einer ADTKD. Eine Wiedervorstellung des Patienten wurde in 4Wochen zur Verlaufskontrolle vereinbart.
Azathioprin wird als Prodrug hepatisch zum wirksamen Metaboliten 6-Mercaptopurin metabolisiert. Dieses wird weiter in 6-Thioguaninnukleotide umgewandelt, welche als falscher Baustein in Nukleinsäure eingebaut werden. Der Metabolit 6-Mercaptopurin wird durch die Xanthinoxidase abgebaut. Eine begleitende Therapie mit Allopurinol hemmt den Abbau von 6-Mercaptopurin zu inaktiver 6-Thioharnsäure. Das Resultat ist wie im vorliegenden Falle eine erhöhte Toxizität durch die erhöhte Bioverfügbarkeit.
Deshalb wird bei gleichzeitiger Einnahme von Allopurinol oder anderen Xanthinoxidasehemmern eine Dosisreduktion auf ein Viertel der Normaldosis empfohlen, um eine erhöhte Hämatotoxizität zu vermeiden. Die häufigste unerwünschte Nebenwirkung ist hierbei die Depression des Knochenmarks mit Leukozytopenie bis hin zur Panzytopenie.
Angelehnt an https://www.krankenhauspharmazie.de/Nebenwirkung-des-Monats/azathioprin.html, Dr. med.A.Knüppel-Ruppert, Prof. Dr. med. H. Dormann, Dr. rer. Nat. B. Pfinstermeister, C. Schnitzer, Dr. med. M. Krag, Klinikum
Aktuelle Medikation:
RAPAMUNE 1 mg überzogene Tabletten 2½ - 0 - 0 - 0
DECORTIN 5 mg Tabletten1 - 0 - 0 - 0
ARANESP 40 µg Inj.-Lsg.i.e. Fert.S.autom.Nadels. Cave Hb über 12mg/dl alle 14 Tage
BENZBROMARON AL 100 überzogene Tabletten 1 - 0 - 0 - 0
IVABRADIN axcount 7,5 mg Filmtabletten 1 - 0 - 1 - 0
ELIQUIS 5 mg Filmtabletten1 - 0 - 1 - 0
PANTOPRAZOL AAA 40 mg magensaftres.Tabletten 1 - 0 - 1 - 0
FOSTER 100/6 µg 120 Hub 2 - 0 - 0 - 2
Dosieraerosol
BICANORM magensaftresistente Tabletten 1 - 0 - 1 - 0
AZOPT 10 mg/ml Augentropfensuspension 1 - 0 - 1 - 0
COTRIMOXAZOL AL 400/80 Tabletten Dienstag/Samstag
DEKRISTOL 20.000 I.E. Weichkapseln alle 14T age
LYRICA 25 mg Hartkapseln 1 - 0 - 1 - 0
FOLSÄURE AL 5 mg Tabletten 1x1/Woche
OLYNTH 0,05% N Schnupfen Dosierspray ohne Konserv.1 Hub - 0 - 1 Hub - 0
NOVAMINSULFON AbZ 500mg/ml Tropfen zum Einnehmen bei Schmerzen bis 4/40Trp.
ATORVASTATIN Heumann 40 mg Filmtabletten 0 - 0 - 1 - 0
EZETIMIB Denk 10 mg Tabletten 0 - 0 - 1 - 0
FERACCRU 30 mg Hartkapseln1 - 1 - 0 - 0
OLMESARTAN Amlodipin1 - 0 - 0 - 0
HCT beta 40mg/5mg/12,5mg FTA unter Kalium- und Kreatininkontrolle
Die Diskussion ob eine Hyperurikämie konsekutiv zu einem erhöhten kardiovaskulären Risiko führt ist kontrovers, da Harnsäure eine nützliche Funktion im Rahmen von Zerstörung von hoch reaktiven schädlichen Sauerstoffradikalen besitzt. Im Gegensatz dazu mehren sich jedoch die Anzeichen dafür, dass eine erhöhte Harnsäure mit einem erhöhten kardiovaskulären Risiko einhergehen könnte. Demzufolge wäre die Harnsäure als unabhängiger prädiktiver Faktor für kardiovaskuläre Mortalität zu sehen.
Da die Hyperurikämie mit dem sogenannten metabolischen Syndrom sowie der arteriellen Hypertonie, Diabetes mellitus und einer Adipositas assoziiert ist, ist es schwierig festzustellen, ob eine Hyperurikämie alleine als kardiovaskulärer Risikofaktor zu interpretieren ist.
In den vergangenen Jahren ist in verschiedenen epidemiologischen Untersuchungen gezeigt worden, dass die Gicht per se ein unabhängiger kardiovaskulärer Risikofaktor zu sein scheint. In dem vorliegenden Fall zeigt sich bei massiver Hyperurikämie ebenfalls eine Assoziation zu Myokardinfarkten und einer KHK. Demzufolge empfehle ich, Patienten mit einer Hyperurikämie im Rahmen einer ADTKD entsprechend ihrem Risikoprofil zu behandeln und die zusätzlichen kardiovaskulären Risikofaktoren zu minimieren und nach Leitlinie zu therapieren (4).
Medikamente zur Harnsäuresenkung bei Nierentransplantation
Febuxostat: Cave: bei bekannten Herzerkrankungen, keine Empfehlung für Organtransplantierte.
Im vorliegenden Fall bei Z.n. Medikamentenintoxikation und Agranulozytose durch die Kombination mit der Gabe von zu hoch dosiertem Azathioprin sind vor einer erneuten Gabe sehr genau Nutzen und Risiko abzuwägen. Obwohl die Kombinationstherapie ursächlich für die Agranulozytose gewesen zu sein scheint, kann diese jedoch auch unter der Monotherapie mit Allopurinol vorkommen. Zudem besteht durch eine Wechselwirkung mit Diuretika (Thiaziden) ein erhöhtes Risiko für ein Steven-Johnson-Syndrom.
Wenn eine erneute Gabe aus Mangel an Alternativen unausweichlich ist, sollte diese nierenfunktionsadaptiert niedrigdosiert und unter engmaschiger Kontrolle des Blutbildes erfolgen.
Benzbromaron ist in diesem Falle das Mittel der Wahl [angelehnt an die Fachinformation].
Patient 2: Sohn von Patient 1
Anamnese der Kindernephrologie des Uniklinikums
Der Patient stellte sich erstmalig mit dem 13. Lebensjahr in der nephrologischen Ambulanz des Universitätsklinikums vor.
In der Anamnese berichtete er darüber, dass mehrere Personen in seiner Familie von einer Hyperurikämie und einige im Erwachsenenalter von einer terminalen Nierenerkrankung betroffen seien.
Bei ihm sei erstmalig mit dem 11. Lebensjahr eine erhöhte Harnsäure gemessen worden. Dieses Ergebnis bestätigte sich zwei Jahre darauf erneut.
Bisher habe der Patient keine schwerwiegenden Erkrankungen gehabt. Die Schwangerschaft, Geburt und Frühklinikum seien regelrecht verlaufen. Im Alter von 7 Jahren sei eine KISS (Kopfgelenk-induzierte Symmetrie-Störung) diagnostiziert und eingerenkt worden. Mittlerweile besucht der Patient die 6.Klasse eines Gymnasiums, die Schulleistungen sind hierbei durchschnittlich. In seiner Freizeit spiele er Fußball und mache Bogenschießen. Allergien oder Unverträglichkeiten seien nicht bekannt. Aufgrund der Erkrankung des Vaters werde eine Diät eingehalten, die Harnsäureexzesse vermeidet.
Familienanamnese:
Siehe Pat. 1
Körperlicher Untersuchungsbefund:
Knapp 13-jähriger Jugendlicher in gutem Allgemeinzustand und schlankem Ernährungszustand, längliches Gesicht, hyperpigmentiert perioral, sonst keine Auffälligkeiten am Integument. Ausgeglichener Hautturgor, keine Ödeme, regelrechter kardiopulmonaler Auskultationsbefund. Bauch weich, kein Druckschmerz, keine Organomegalien, keine Klopfschmerzen über der Wirbelsäule oder im Nierenlager. HNO-Bereich infektfrei, keine pathologischen Lymphknoten zervikal oder axillär.
Gewicht 43 kg (34. Perzentile), Länge 158,1 cm (48. Perzentile), BMI 17,2 kg/m2 (28. Perzentile), RR 109/60 mmHg; Puls 66 die Minute.
Sonografie der Nieren:
Altersentsprechender Normalbefund.
Beurteilung:
Bei dem Patienten konnte wie bereits anamnestisch und klinisch angenommen die Diagnose einer familiären juvenilen hyperurikämischen Nephropathie molekulargenetisch gesichert werden. Es liegt ein autosomal-dominanter Vererbungsvorgang vor, wie auch die Familienanamnese eindrucksvoll belegt. Hierbei zeigt sich die GFR leicht reduziert mit 72ml/min pro KOF. Es besteht eine wenig ausgeprägte Hyperurikämie bei niedrig fraktionierter Urea-Exkretion.
Insbesondere in Anbetracht der deutlich erhöhten kardiovaskulären Morbidität bei den betroffenen Familienmitgliedern empfiehlt sich der Beginn einer harnsäuresenkenden Therapie. Der Blutdruck des Patienten zeigte sich in ambulanten Einzelmessungen normotensiv.
In der anschließenden Vorstellung in der Stoffwechselambulanz der Universitätsklinik konnte ein normwertiger Purin- und Pyrimidinstoffwechsel diagnostiziert werden. Die Harnsäurewerte waren bei dem Patienten erhöht, daher wurde eine Therapie mit Benzbromaron begonnen. Nebenbefundlich zeigt sich ein deutlicher Vitamin-D-Mangel bei sekundärem Hyperparathyreoidismus. Hier wurde eine entsprechende Substitution begonnen.
Aktuelle Medikation:
BENZBROMARON AL 100 überzogene Tabletten 1 - 0 - 0 - 0
DEKRISTOL 1.000 I.E. Tabletten1 - 0 - 0 - 0
Labor:
Harnsäure 9,97mg/dl erhöht (Normwert 3,1 – 7,0)
Diagnosen:
- Familiäre juvenile hyperurikosämische Nephropathie (FJHN, autosomal-dominant) mit heterozygoter UMOD-Mutation (c. 1463 G > A)
- Leicht eingeschränkte Nierenfunktion, CKD 2 nach KDIGO (GFR 65ml/min/1,73 m² KOF)
- Sekundärer Hyperparathyreoidismus mit Vitamin-D-Mangel
ADTKD im konkreten Fall der Patientenbeispiele
Typisch an den beiden Fällen ist das Auftreten einer Hyperurikämie in jungen Jahren und die unterschiedliche Manifestation einer chronisch funktionellen Nierenerkrankung mit Progression zur terminalen, dialysepflichtigen Nierenerkrankung in der Familie. Die Mutter der acht Geschwister verstarb mit dem 46. Lebensjahr an einer "Gichtniere" im Rahmen eines Coma urämicum bei begleitendem klinischen Bild einer exazerbierten Gicht mit Gichtablagerungen in der Haut und Ulzerationen.
Eine Tochter verstarb rasch nach der Geburt, erreichte somit nicht das Alter einer klinischen Manifestation einer ADTKD. Die zweite Tochter wurde mit ca. 30 Jahren dialysepflichtig und nierentransplantiert. Ein Sohn erreichte im 52. Lebensjahr das Stadium einer dialysepflichtigen Nierenerkrankung bei Rheuma und Gichtniere und wurde demzufolge nierentransplantiert. Er litt begleitend unter einer KHK.
Ein weiterer Sohn wurde im Alter von 42 Jahren dialysepflichtig und erhielt eine Nierentransplantation.
Der dritte Sohn erreichte durch seinen Unfall nicht das Alter einer klinischen Manifestation der ADTKD. Die Penetranz in der Familie dieser UMOD-Mutation bleibt somit unklar. Die restlichen Familienmitglieder befinden sind nach wie vor im weiten Raster einer mögliche Dialysepflichtigkeit zwischen dem 20. und 80.Lebensjahr. Die Familie entspricht somit vielen anderen ADTKD-UMOD-Mutationspatienten.
In der nächsten Generation wurde dann bei dem Sohn des im 42. Lebensjahr Nierentransplantierten eine genetische Untersuchung mittels NextGenerationSequencing durchgeführt und erstmalig die Diagnose einer autosomal-dominanten tubulointerstitiellen Nierenerkrankung im Rahmen einer UMOD-Mutation diagnostiziert. Die Familienanamnese mit ihren beiden Patientenbeispielen ist in diesem Fall beispielhaft für die Erkrankung.
- Autosomal-dominante genetische Mutation
- Progressiver Funktionsverlust der Niere
- Blandes Urinsediment
- Abwesenheit von Proteinurie und Albuminurie
- Keine arterielle Hypertonie im Frühstadium
- In der Anamnese keine medikamentöse Therapie, die eine tubulointerstitielle Nephritis erklären könnte
- Normale Nierengrößen im Ultraschall, im Verlauf kommen vermehrt Schrumpfnieren vor
- Nykturie oder Enuresis bei Kindern (Verlust von renaler Konzentrationsfähigkeit)

Auf unserem CME-Portal www.kirchheim-forum-cme.de können Sie ab dem 29.08.2023 diesen Beitrag bearbeiten und bekommen bei Erfolg Ihre Punkte sofort gutgeschrieben.
Erschienen in: Der Nierenarzt, 2023; 10 (4) Seite 10-20